5. The assemble module¶
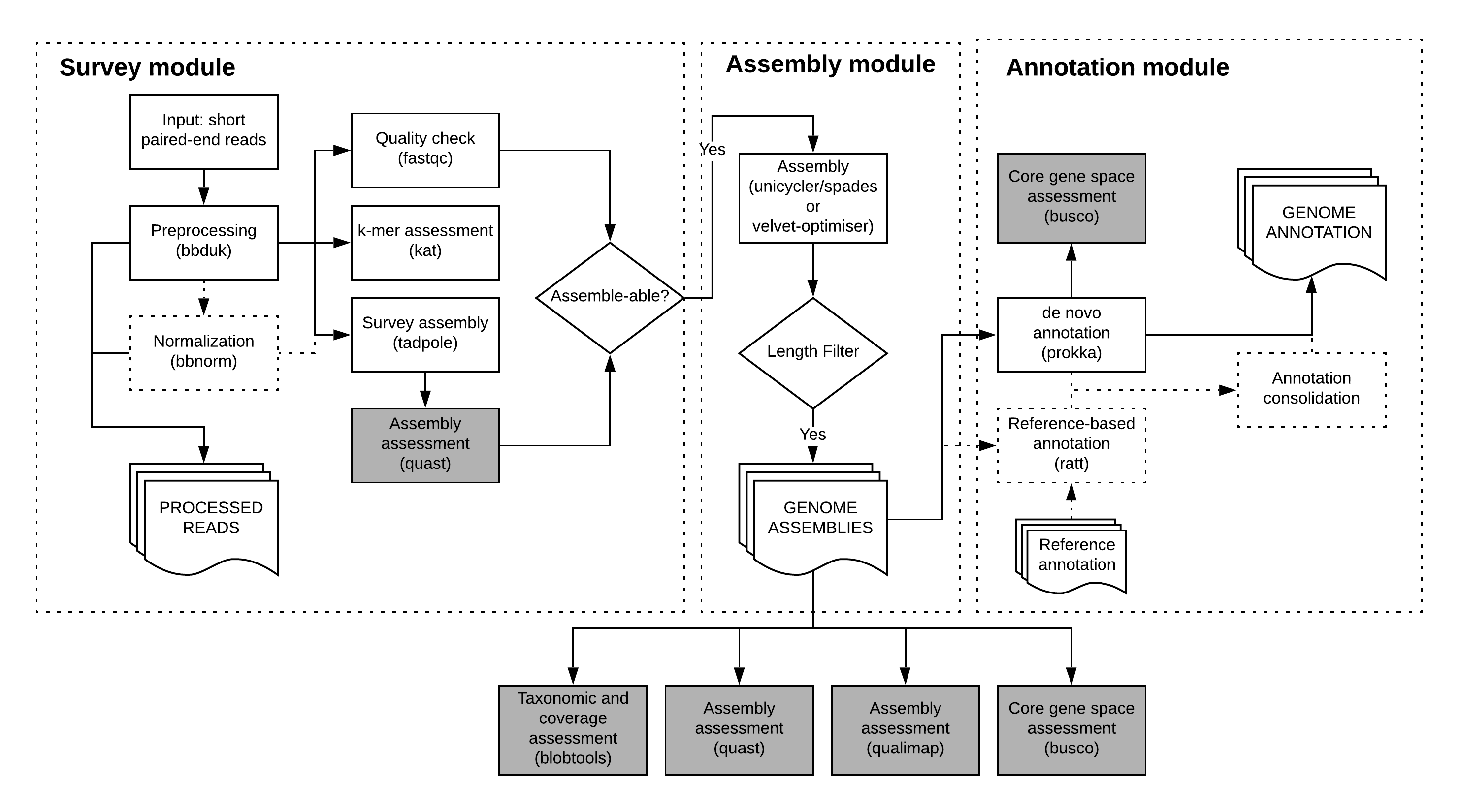
The assemble module performs automatically optimized assemblies of the provided library data.
5.1. Sample sheet preparation¶
If the user has previously run the survey module, the resulting samplesheet samplesheet.qc_pass.tsv can be used directly
to drive the assemble module.
Otherwise, the user should prepare a comma-separated samplesheet following the column order below.
- Sample ID
- Sample Name (can be the same as Sample ID; the intention is to allow a more understandable sample reference in the future)
- Full path to R1 file
- Full path to R2 file
- LEAVE EMPTY (intended use: Full path to single-end file)
- LEAVE EMPTY (intended use: NCBI taxonomy id)
- LEAVE EMPTY (intended use: Taxonomy name)
- LEAVE EMPTY (intended use: FastQC report for raw R1)
- LEAVE EMPTY (intended use: FastQC report for raw R2)
- LEAVE EMPTY (intended use: FastQC report for raw single-end file)
- Full path to preprocessed, non-normalized R1 file
- Full path to preprocessed, non-normalized R2 file
- LEAVE EMPTY (intended use: Full path to preprocessed, non-normalized single-end file)
- Full path to preprocessed, normalized R1 file
- Full path to preprocessed, normalized R2 file
- LEAVE EMPTY (intended use: Full path to preprocessed, normalized single-end file)
5.2. Command line arguments¶
The command bgrrl -h or bgrrl <stage> -h (or --help instead of -h) will display a list of command line options.
Remember each bgrr| run requires at the very least the following three command line parameters:
input_sheet--config--hpc_config
5.2.1. assemble options:¶
--assembler {unicycler,velvet}Assembly software to use for genome assembly. [unicycler]
--contig-minlen CONTIG_MINLENMinimum length [bp] of contigs retained in filtering step [0].
--no-normalizationUse non-normalized reads in assemble module [False]
--run-annotation- Run annotation on assembly. If set, de novo annotation
with prokka will be run. Additionally, you may enable annotation transfer by specifying a path to a reference annotation with
--ratt-reference. [False]
--custom-prokka-proteins CUSTOM_PROKKA_PROTEINSIf you have a custom protein database that you would like prokka to use (
prokka’s--proteinsoption), then specify the path to it here. [n/a]--ratt-reference RATT_REFERENCEPath to reference data for ratt annotation transfer
--is-final-step- If set, analysis packaging will take place after the
assembly stage. Otherwise, assume that an annotation stage will follow, which will then take care of analysis packaging. [False]
--no-packagingDisable automatic packaging. [False]
--prokka-package-style {by_sample,all_in_one}Should the prokka annotation be packaged into one directory per sample (by_sample) or into one single directory (all_in_one)? [by_sample]